Inicio / Archivo / Año 2, No 2, septiembre 2019 - agosto 2020 / Paper 10

EL DISEÑO DE FÁRMACOS ASISTIDO POR COMPUTADORA. ¿MITO O REALIDAD?
Ana María Velázquez-Sánchez*, Víctor Hugo Vázquez-Valadez, Manuel Alejandro Hernández-Serda, Pablo Aguirre-Vidal y Enrique Ramón Ángeles-Anguiano
Facultad de Estudios Superiores Cuautitlán UNAM
anamvs@unam.mx
Resumen
El descubrimiento de los fármacos ha evolucionado desde el estudio de la “medicina tradicional”, en donde los primeros hallazgos de sustancias activas se llevaron a cabo por serendipia y su difusión mediante tradición oral. Actualmente, el desarrollo de nuevas sustancias activas se lleva a cabo con base en la relación estructura química y actividad biológica empleando inteligencia artificial, es decir, se desarrolla el diseño de nuevos fármacos asistido por computadora (conocido en español con la sigla DIFAC, del inglés Computer-Aided Drug Design). Para el diseño de nuevas estructuras con diversas actividades biológicas, en este trabajo se usaron métodos QSAR/CoMFA en 2 y 3 dimensiones (del inglés Quantitative Structure-Activity Relationship, relación cuantitativa estructura-actividad y Comparative Molecular Field Analysis, análisis comparativo de campo molecular), acoplamiento molecular o docking y dinámica molecular empleando programas computacionales como AutoDock, Sybyl y MOE (del inglés Molecular Operating Environment, entorno operativo molecular). Empleando el diseño de fármacos asistido por computadora en la Facultad de Estudios Superiores Cuautitlán UNAM se han diseñado fármacos antihipertensivos, antiarrítmicos, anticancerígenos e ixodicidas.
Palabras clave: Análisis conformacional, optimización geométrica, dinámica molecular, QSAR, CoMFA, docking.
Introducción
El diseño de fármacos asistido por computadora (DIFAC, del inglés Computer-Aided Drug Design) es un área del diseño racional de fármacos (Boyd, 1999) y consiste en aplicar algún procedimiento realizado por una computadora para relacionar la actividad biológica de un compuesto con las propiedades de su estructura (Hopfinger, 1985).
Este tipo de diseño es una herramienta que además incluye estudios estadísticos de las propiedades de la estructura y se emplea para obtener moléculas con un porcentaje elevado de la actividad biológica requerida.
El DIFAC ha sido impulsado por el gran avance que se ha tenido en las Ciencias de la Computación, actualmente se cuenta con máquinas construidas con procesadores más veloces y programas computacionales que dan resultados con mayor precisión en los estudios de las estructuras químicas moleculares (Balbes, Mascarella, & Boyd, 1996; Shen et al., 2004; Sippl, 2002).
Este diseño se emplea para formalizar, eficientar y obtener fármacos de manera racional, además, permite una mejor descripción del proceso biológico in sílico para que posteriormente sea comprobado mediante experimentos in vitro o in vivo. Estos procesos de validación son biyectivos, es decir, los estudios in sílico también se llevan a cabo después de tener conclusiones de los experimentos in vitro e in vivo.
El DIFAC se lleva a cabo mediante 2 metodologías generales: 1) estudios vía receptor y 2) estudios vía ligando (Tropsha & Zheng, 2001).
El diseño basado en la estructura del receptor incluye estudios por homología, estudios docking (acoplamiento molecular) y dinámica molecular, entre otros.
El diseño mediante la estructura del ligando incluye: estudios de la relación estructura–actividad biológica en 2 dimensiones (QSAR 2D), estudios de relación estructura–actividad biológica en 3 dimensiones (QSAR 3D) tales como CoMFA (Comparative Molecular Fields Analysis, análisis comparativo de campo molecular), CoMSIA (Comparative Molecular Similarity Indices Analysis, análisis comparativo de índices de similitud molecular), etc., todos ellos basados en estudios de la estructura química molecular y estudios estadísticos.
Ya sea que se utilice un tipo de método u otro, la finalidad es la predicción de la actividad biológica o de alguna otra propiedad farmacológica o fisicoquímica, de una serie de moléculas.
La siguiente etapa consiste en sintetizar las moléculas seleccionadas como mejores candidatos y realizar las evaluaciones biológicas y/o fisicoquímicas correspondientes.
Todos los métodos de diseño por computadora son validados experimentalmente mediante diversas técnicas espectroscópicas tales como: Difracción de rayos X, resonancia magnética nuclear y espectroscopía infrarroja, entre otras. Las estructuras de los receptores se obtienen de bases de datos gratuitas.
Algunos fármacos que, mediante el DIFAC, han llegado a ser medicamentos empleados actualmente son: norfloxacina (antibacteriano), losartán (antihipertensivo), dorzolamida (glaucoma), nelfinavir (sida), eprosartan (antihipertensivo), zolmitriptan (migraña), indinavir (antiviral, sida), captopril (antihipertensivo), entre muchos otros (Stout, Matier, Barcelon-Yang, Reynolds, & Brown, 1985).
Los fármacos que se han diseñado en la FES Cuautitlán son:
- LQM 319 y LQM 337 (LQM, sigla de Laboratorio de Química Medicinal) con actividades antihipertensivas y antiarrítmicas, cuyo receptor es la enzima convertidora de angiotensina (ECA); para estos compuestos se tomó como base al compuesto de origen chino denominado changrolina (Stout, et al., 1985).
- Se han desarrollado también compuestos con actividad quimio-protectora como los compuestos análogos del CAPE (del inglés Caffeic Acid Phenethyl Ester, éster fenetílico del ácido cafeico) (Carrasco-Legleu et al., 2006).
- También se han diseñado una serie de ixodicidas entre los que destacan el (4-bromofenil) carbamato de etilo (LQM 919) y el (4-clorofenil) carbamato de etilo (LQM 996), cuya actividad biológica está dirigida a receptores tales como: tubulina alfa y beta, estos ixodicidas afectan el proceso de oviposición de las garrapatas Rhipicephalus microplus (Prado-Ochoa et al., 2014).
A continuación, se desarrollará brevemente, la metodología para la obtención de los antihipertensivos y antiarrítmicos, derivados morfolínicos y tiomorfolínicos de fenol.
Objetivo
Aplicar el diseño asistido por computadora para proponer nuevos compuestos morfolínicos y/o tiomorfolínicos del fenol que presenten actividad biológica como antihipertensivos y antiarrítmicos para sintetizar y probar a los mejores candidatos e ir conformando en lo posible las series de entrenamiento y prueba, además de diseñar al menos 2 nuevos compuestos con dicha actividad empleando el DIFAC.
Metodologías
La metodología de trabajo se describe por cada actividad realizada en el proyecto. Solo ejemplificaremos 2 actividades de las 6 que se llevaron a cabo:
- Síntesis y purificación de 27 compuestos derivados morfolínicos y tiomorfolínicos de fenol empleando 2 fuentes diferentes de energía para las reacciones. La síntesis de los diversos derivados de (metil–il–morfolín) fenol y (metil–il–tiomorfolín) fenol se llevó a cabo por los métodos de sustitución electrofílica aromática (SEA) y SEA vía base de Mannich (SEA_BM), en cada método lo que cambió es el orden de mezclado de los reactivos, dos fuentes diferentes de transferencia de energía a la reacción, el primero fue a reflujo y el segundo con radiación infrarroja en ausencia de disolventes (Velázquez et al., 2006). Después de la síntesis, se llevó a cabo la caracterización espectroscópica incluyendo la difracción de rayos X de las estructuras químicas de cada compuesto.
- Los estudios teóricos que se llevaron a cabo son los siguientes: a) Análisis conformacional empleando el paquete Systematic Conformational Search del programa de modelado molecular Sybyl versión 6.9 (Dunbrack, 2004). La determinación de la energía potencial de cada molécula de los compuestos LQM 300 se realizó mediante una búsqueda sistemática, rotando los enlaces. Se inició con incrementos de rotación de los enlaces de 30 °. b) Optimización geométrica y cálculo de la energía empleando el método semiempírico PM3 se efectuaron con el paquete MOPAC del programa en modelado molecular Sybyl versión 6.9 (Dunbrack, 2004). La estructura del compuesto en estudio se tomó del mínimo conformacional obtenido del análisis conformacional. c) Métodos ab initio. La optimización geométrica y el cálculo de energía se realizaron empleando el paquete NWChem versión 4.5 (Northwest Computacional Chemistry) (Andrés & Beltrán, 2001; Atkins & Friedman, 1997; Bylaska et al., 2004; Karelson, 2000). d) Método Funcionales de la Densidad. La optimización geométrica y el cálculo de energía se realizaron empleando el mismo paquete que en el inciso anterior. Los conjuntos de base empleados para el cálculo fueron: B3LYP/DZVP/A1//B3LYP/DZVP/A1 y B3LYP/6-31g// B3LYP/6-31g (Andrés & Beltrán, 2001; Atkins & Friedman, 1997; Bylaska et al., 2004; Cambridge Sort Corporation, 2003; Karelson, 2000). e) Validación del método teórico. La validación del método teórico que describe mejor la estructura de los compuestos a estudiar consistió en comparar los resultados de la optimización geométrica obtenida por la aplicación de los diversos métodos con la estructura geométrica resultante del análisis estructural realizado por difracción de rayos X. Otro método de validación consistió en realizar la superposición de las estructuras experimentales de rayos X con las estructuras resultantes de la optimización de cada método teórico empleado con el programa Sybyl y evaluar el % de desviación estándar de la sobreposición de estas (Dunbrack, 2004). f) Determinación de descriptores electrónicos, estéricos y fisicoquímicos. Una vez validado el método, se eligen las estructuras resultantes y se calculan teóricamente los siguientes descriptores electrónicos: E. HOMO (del inglés Highest Occupied Molecular Orbital, Orbital Molecular Ocupado más Alto) (eV), E. LUMO (del inglés Lowest Unoccupied Molecular Orbital, Orbital Molecular No Ocupado más Bajo) (eV), fórmula molecular, simetría, energía total, MD (Momento Dipolar, en Debyes), E. electrónica. También se calcularon los descriptores estéricos: Índice de Balaban, Área de Connolly, Índice de Wiener, Volumen, Área, Refractividad molar, Log P y los descriptores fisicoquímicos: E. de vibración cero, ΔH de formación, E. vdW no debida a las interacciones de los átomos -1,4, E. vdW -1,4 y E. de solubilidad en agua, aplicando un “single point” a la estructura optimizada con el programa Chem 3D® ULTRA, versión 8.03 (Andrés & Beltrán, 2001; Atkins & Friedman, 1997; Bylaska et al., 2004; Cambridge Sort Corporation, 2003; Karelson, 2000).
Resultados
Algunos ejemplos de los resultados se mostrarán en las siguientes tablas y figuras para algunas de las actividades.
Tabla 1. Resumen de rendimientos, tiempos de reacción y metodologías de síntesis. | ||||
LQM | Reacción | Rendimiento | Punto fusión/ ebullición (° C) | Tiempo de reacción |
301 | SEA_BM // IR | 17.71 % // 83.61 % | 188 -190 | 3 h 45’ // 54’ |
302 | SEA_BM // IR | 34.78 % | 156 -158 | 5 días // 17’ |
303 | SEA_BM // IR | 50.53% | 248 – 250 (eb.) | 5 días // 20’ |
304 | SEA // IR | 5.11% // 58.0% | 75 - 78 | 56 horas // 25’ |
305 | SEA // IR | 37.55% // 57.44% | 129-131 | 5.0 h // 25’ |
306 | SEA // IR | 82.0% // 92.0% | 114 - 116 | 6 horas // 12’ |
307 | SEA // IR | 78.0% // 85.0% | 102 - 104 | 4.0 h // 23’ |
308 | SEA // IR | // 90.0% | 95-97 | 31 h // 32’ |
309 | SEA // IR | 20.0% // 95.0% | 124 - 126 | 15 h // 9.0’ |
310 | SEA // IR | NA // 90.0% | 202 – 204 | 8.0 h // 28’ |
311 | SEA // IR | 70.0% | 114 – 116 | 15’ // 8’ |
312 | SEA // IR | 85.0% // 95.0% | 108 – 110 | 36.0 h // 32’ |
313 | SEA // IR | 83.0% | 78 – 80 | 36.0 h // 40’ |
314 | SEA // IR | 87.0% // 87.0% | 104 -106 | 38.0 h // 21’ |
315 | SEA_BM | 84.0 | 8 días | |
316 | SEA | 113 | 3 días | |
317 | SEA // IR | 25.0 // 60.0% | 127 – 129 | 24.0 h // 7.0’ |
318 | SEA // IR | 2.11% // 70.0% | 85 – 87 | 48.0 h // 9.0’ |
319 | SEA // IR | 34.55% // 80.0% | 95 – 97 | 48.0 h // 15.0’ |
320 | SEA | 11.92% | 163 – 165 | 47.0 h 30’ |
321 | SEA | 0.98% | 136 – 138 | 47.0 h 30’ |
322 | SEA // IR | 83.31% // 85.0% | 181 -183 | 6.0 h // 6.0’ |
323 | SEA | 87.65% | 193-195 | 36.0 h |
324 | SEA // IR | 30.25 % // 38.0% | 112 – 114 | 20 d 22.25 h //10’ |
Tabla 2. Respuesta biológica de algunos compuestos LQM. | |||
Compuestos | ΔPMÁX sistólica/tiempo (mmHg/min) | ΔPMÁX diastólica/tiempo (mmHg/min) | ΔMÁX Frecuencia cardiaca /tiempo (latidos/min/min) |
LQM 301 | 73.9 / 100 | 46.7 / 100 | 72.2 / 110 |
LQM 318 | 71.47 / 100 | 31.8 / 60 | 147.88 / 100 |
LQM 319 | 95.755 / 90 | 48.225 / 90 | 160.448 / 100 |
LQM 322 | 45.6 / 60 | 29.2 / 60 | 92.5 / 120 |
LQM 320 | 28.086 / 60 | 12.5875 / 60 | 106.566 / 120 |
CAPTOPRIL | 42.0 / 50 | 36.45 / 100 | 52.04 / 70 |
LOSARTÁN | 82.3 / 80 | 66.35 / 80 | 38.37 / 80 |
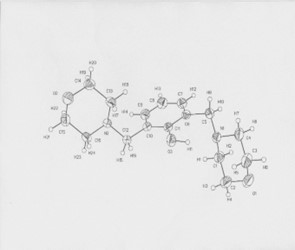
Figura 1. Resultado del análisis de difracción de rayos X del compuesto LQM 309..
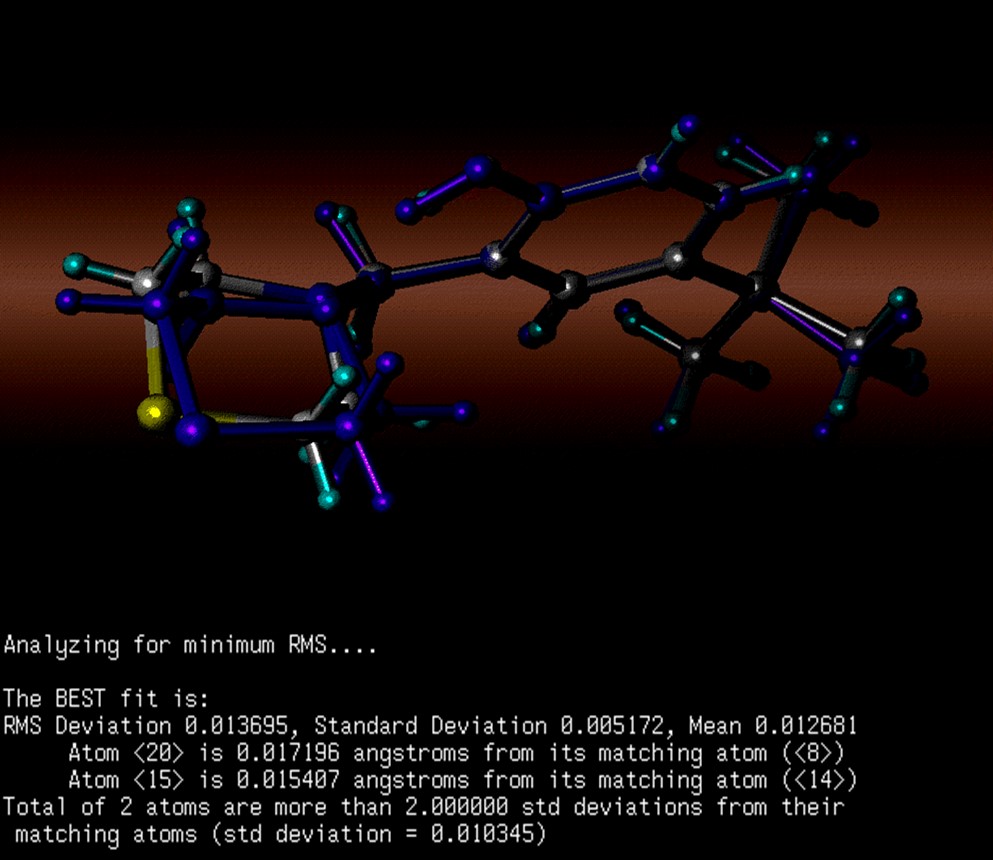
Figura 2. Comparación de la estructura obtenida por el método DFT (B3lyp/6-31g) con la estructura obtenida por análisis rayos X del LQM 318.
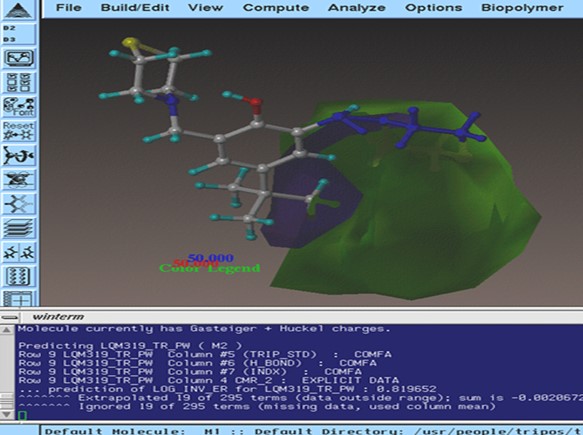
Figura 3. Predicción de la actividad antiarrítmica del compuesto LQM 319.
Discusión
En la Tabla 1 se muestra que cuando la transferencia de calor a las reacciones se lleva a cabo mediante radiación IR, en general, los tiempos de reacción disminuyen y los rendimientos de las reacciones aumentan, lo cual es muy importante en el trabajo de síntesis de fármacos.
En la Tabla 2 se comparan las actividades biológicas de algunos compuestos y se observa que el LQM 319 (resultado de emplear DIFAC) es similar al de Losartán que tiene un efecto tanto en la presión arterial sistólica y diastólica, no así en la frecuencia cardiaca en el modelo de rata hipertensa espontánea empleado para esta prueba.
Las Figuras 1 y 2 están relacionadas con la validación de los métodos de los cálculos teóricos que se emplearon para la optimización geométrica de las estructuras. Primero se muestra, como ejemplo, los resultados de difracción de rayos X para la estructura del compuesto LQM 309, estas caracterizaciones se hicieron para toda la serie de compuestos que se sintetizaron. Estos datos sirven para compararlos computacionalmente y ver el grado de correlación entre la estructura experimental y la obtenida mediante los cálculos teóricos y definir los métodos de cálculos a emplear.
Finalmente, la Figura 3 es solo un ejemplo de cómo se obtiene la predicción de la actividad biológica de la estructura haciendo el análisis de los compuestos en cuanto a sus campos estéricos y electrostáticos, entre otros parámetros fisicoquímicos.
Este trabajo constituye el primer estudio preliminar de los compuestos LQM-300’s que mostraron tener actividad como hipotensores y disminución de la frecuencia cardiaca (en algunos casos) en modelos de rata anestesiada normotensa y de rata hipertensa espontánea consciente.
Conclusión
Aplicando el DIFAC basado en la estructura del ligando en 3D, se propusieron dos nuevos compuestos LQM 319 y LQM 337 derivados tiomorfolínicos de fenol de los cuales se predijo su actividad biológica y que experimentalmente mostraron tener efecto de disminución sobre la presión sistólica, presión diastólica y algunos, sobre la frecuencia cardiaca empleando una serie de entrenamiento de 25 compuestos derivados morfolínicos y tiomorfolínicos de fenol y una serie de prueba de ocho compuestos de entre los 25 para el estudio CoMFA, dichas series se formaron respetando el esqueleto básico del compuesto líder. Los compuestos fueron sintetizados vía base de Mannich empleando la radiación de luz infrarroja como fuente de energía lo cual redujo notablemente los tiempos de síntesis e incrementó los rendimientos de las reacciones.
Las siguientes conclusiones son para cada uno de los objetivos que se marcaron para este trabajo (no mostrados en los resultados) y son un ejemplo del alcance del DIFAC: En general los mínimos conformacionales, encontrados en los estudios de mecánica molecular, corresponden a los rotámeros cuyos anillos con los heteroátomos se encuentran en las posiciones cis y trans al plano del fenol. El rotámero cis es el de menor energía, aunque la estructura del rotámero trans se encontró en el análisis por difracción de rayos X de algunos de los compuestos, lo cual puede deberse en gran medida a las interacciones intermoleculares e intramoleculares involucradas en la formación de los cristales.
En los estudios de química cuántica, el método ab initio empleando el conjunto de base 6-31 g sin difusión es el que mejor describe las estructuras encontradas en el análisis por difracción de rayos X de los compuestos en estudio.
Los compuestos LQM de la serie 300 mostraron tanto efecto de disminución sobre la frecuencia cardiaca como actividad hipotensora en los modelos de rata anestesiada y de rata hipertensa espontánea. El modelo obtenido en los estudios CoMFA, en donde se relacionaron los campos estéricos, electrostáticos, la refractividad molar calculada y la refractividad molar calculada al cuadrado con el efecto de disminución de la frecuencia cardiaca, predijo una actividad aceptable para los compuestos diseñados LQM 319 y LQM 337, de manera que, al observar los datos reportados para los estudios de etapa I en el modelo de rata hipertensa, el efecto del compuesto LQM 319 fue mayor que el de losartán en la presión sistólica, menor en la presión diastólica y mayor en el efecto presentado sobre la frecuencia cardiaca, así mismo, dicho efecto fue mayor que el de los demás compuestos de la serie LQM-300’s estudiados en ese modelo, esto es notable, ya que se demuestra que se puede llevar a cabo el estudio CoMFA de la serie de prueba de estos compuestos, aunque el valor de q2 obtenido sea menor a 0.5 porque el porcentaje de error relativo encontrado en la predicción es aceptable.
Se propone que los estudios preliminares tanto de evaluación del efecto hipotensor como del efecto de disminución de la frecuencia cardiaca se diseñen y se validen estadísticamente para confirmar o modificar los resultados en la primera etapa del estudio.
Agradecimientos
Al Consejo Nacional de Ciencia y Tecnología (CONACyT) y a la DGAPA. A la Coordinación del Posgrado en Ciencias Químicas. A los proyectos PAPIIT IN213606 e IN207705 y a la Cátedra IN 1-11 de la FESC. A Dr. José Luis Medina Franco, Dr. Rafael Castillo, Dra. Ma. de Jesús Rosales e Ing. Marco Antonio Leyva (laboratorio de Rayos X del CINVESTAV), MC. Rosa Isela del Villar, MC. Oscar Yánez y MC. Georgina Duarte (del USAI), Dra. Luisa Martínez (del laboratorio de Farmacología del Miocardio de la FESC), Dr. César Compadre (de la Universidad de Arkansas), Dr. Jesús Jiménez Barbero y Dr. Javier Canales (ambos del CIB del CSIC, Madrid).
Referencias
- Andrés, J., & Beltrán, J. (2001). Química teórica y computacional. Publicacions de la Universitat Jaume I. Recuperado de https://books.google.com.mx/books?id=EX4WgX1VgOoC
- Atkins, P. W., & Friedman, R. (1997). Molecular Quantum Mechanics (3a ed.). Oxford University Press. Recuperado de https://books.google.com.mx/books?id=bV5OPgAACAAJ
- Balbes, L. M. ., Mascarella, S. W., & Boyd, D. B. (1996). A perspective of modern methods in computer-aided drug design (Vol. 39, pp. 1568–1569). https://doi.org/10.1021/jm960033i
- Boyd, D. B. (1999). Is rational design good for anything? ACS Symposium Series, 719. https://doi.org/10.1021/bk-1999-0719.ch022
- Bylaska, E., de Jong, W., Govind, N., Kowalski, K., Straatsma, T. P., Valiev, M., … Hammond, J. (2004). NWChem, A Computational Chemistry Package for Parallel Computers, Version 4.5. Pacific Northwest National Laboratory, Richland, Washington, 99352, 999.
- Cambridge Sort Corporation. (2003). Chem 3D ULTRA. Molecular Modeling and Analysis. Cambridge Sort Corporation.
- Carrasco-Legleu, C. E., Sánchez-Pérez, Y., Márquez-Rosado, L., Fattel-Fazenda, S., Arce-Popoca, E., Hernández-García, S., & Villa-Treviño, S. (2006). A single dose of caffeic acid phenethyl ester prevents initiation in a medium-term rat hepatocarcinogenesis model. World Journal of Gastroenterology, 12(42). https://doi.org/10.3748/wjg.v12.i42.6779
- Dunbrack, R. L. (2004). SYBYL. En Dictionary of Bioinformatics and Computational Biology. https://doi.org/10.1002/0471650129.dob0722
- Hopfinger, A. J. (1985). Computer-Assisted Drug Design. Journal of Medicinal Chemistry, 28(9). https://doi.org/10.1021/jm00147a001
- Karelson, M. (2000). Molecular descriptors in QSAR/QSPR.
- Prado-Ochoa, M. G., Ramírez-Noguera, P., Díaz-Torres, R., Garrido-Fariña, G. I., Vázquez-Valadez, V. H., Velázquez-Sánchez, A. M., … Alba-Hurtado, F. (2014). The action of two ethyl carbamates on acetylcholinesterase and reproductive organs of Rhipicephalus microplus. Veterinary Parasitology, 199(3–4). https://doi.org/10.1016/j.vetpar.2013.10.028
- Shen, M., Béguin, C., Golbraikh, A., Stables, J. P., Kohn, H., & Tropsha, A. (2004). Application of Predictive QSAR Models to Database Mining: Identification and Experimental Validation of Novel Anticonvulsant Compounds. Journal of Medicinal Chemistry, 47(9). https://doi.org/10.1021/jm030584q
- Sippl, W. (2002). Development of biologically active compounds by combining 3D QSAR and structure-based design methods. Journal of Computer-Aided Molecular Design, 16(11). https://doi.org/10.1023/A:1023888813526
- Stout, D. M., Matier, W. L., Barcelon-Yang, C., Reynolds, R. D., & Brown, B. S. (1985). Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 3. Modifications to the linkage region (region 3). Journal of Medicinal Chemistry, 28(3), 295–298. https://doi.org/10.1021/jm00381a006
- Tropsha, A., & Zheng, W. (2001). Computer Aided Drug Design. En Computational Biochemistry and Biophysics. https://doi.org/10.1201/9780203903827
- Velázquez, A. M., Torres, L. A., Díaz, G., Ramírez, A., Hernández, R., Santillán, H., … Angeles, E. (2006). A novel one pot, solvent-free Mannich synthesis of methylpiperidinyl phenols, methylphenylmorpholinyl phenols and methylthiophenylmorpholinyl phenols using infrared light irradiation. Arkivoc, 2006(2). https://doi.org/10.3998/ark.5550190.0007.218
Segundo Congreso Nacional de Tecnología 6, 7 y 8 de noviembre de 2019.
Centro de Asimilación Tecnológica y Vinculación.
Facultad de Estudios Superiores Cuautitlán - UNAM

D. R. © UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO.
Excepto donde se indique lo contrario esta obra está bajo una licencia Creative Commons Atribución No comercial, No derivada, 4.0 Internacional (CC BY NC ND 4.0 INTERNACIONAL).
https://creativecommons.org/licenses/by-nc-nd/4.0/deed.es

ENTIDAD EDITORA
Facultad de Estudios Superiores Cuautitlán.
Av. Universidad 3000, Universidad Nacional Autónoma de México, C.U., Delegación Coyoacán, C.P. 04510, Ciudad de México.
FORMA SUGERIDA DE CITAR:
Velázquez-Sánchez, A. M., Vázquez-Valadez, V. H., Hernández-Serda, M. A., Aguirre-Vidal, P., y Ángeles-Anguiano, E. R. (2019). El diseño de fármacos asistido por computadora. ¿mito o realidad?. MEMORIAS DEL CONGRESO NACIONAL DE TECNOLOGÍA (CONATEC), Año 2, No. 2, septiembre 2019 - agosto 2020. Facultad de Estudios Superiores Cuautitlán. UNAM. https://tecnicosacademicos.cuautitlan.unam.mx/CongresoTA/memorias2019/mem2019_paper50.html