Inicio / Archivo / Año 4, No 4, septiembre 2021 - agosto 2022 / Paper 02

INCIDENCIA DE RASGO DREPANOCITICO EN PACIENTES DE
CENTRO DIAGNOSTICO HATVER EN BOCA DEL RIO, VERACRUZ
Jesus Adriana Peres-Quintal, Edith Monserrat García-Carrera y
David Augusto Hatchett-Arenas.
Universidad Veracruzana
*adryquintal@gmail.com
Resumen
Los glóbulos rojos contienen aproximadamente 500-600 millones de moléculas de hemoglobina, en el caso del adulto la que predomina es la hemoglobina A (HbA) formada por 4 cadenas polipeptídicas, 2 cadenas alfa y 2 cadenas beta, además del grupo hem formado por hierro y protoporfirina. Los defectos en la molécula de hemoglobina o hemoglobinopatías pueden ser alteraciones en la síntesis completa o parcial de las cadenas de hemoglobina, en la cual su estructura química está controlada genéticamente; la patología recibe el nombre según la cadena en la que se encuentre el déficit. En el caso de la hemoglobina S hay una sustitución en el aminoácido número 6 de una de las cadenas beta, en el cual el ácido glutámico es sustituido por valina, modificando así su estructura y por lo tanto alterando el organismo que porta este defecto. El rasgo drepanocítico ocurre cuando una persona hereda un gen de hemoglobina S de uno solo de los padres, siendo portadores heterocigotos de la enfermedad; a diferencia de la enfermedad drepanocítica o anemia de células falciformes, en la cual son heredados dos genes de hemoglobina S, uno de la madre y uno del padre, siendo así pacientes homocigotos provocándoles graves consecuencias en su salud. La mayoría de los pacientes con rasgos drepanocíticos son portadores asintomáticos, o presentan síntomas leves de la enfermedad, suelen ser sintomáticos en situaciones de hipoxia; y es por eso de suma importancia identificar a tiempo la presencia de esta alteración en la molécula de hemoglobina, para evitar así una posible anemia de células falciformes en los hijos de los portadores con las posibles complicaciones que presenta la enfermedad.
Palabras clave: anemia, hemoglobina S, inducción de drepanocitos, portadores.
Introducción
La hemoglobina humana es una metaloproteína que posee una estructura cuaternaria, derivada de la formación de 4 subunidades de proteínas globulares.
La importancia de las hemoglobinopatías en el ámbito de la salud pública ha suscitado un acelerado desarrollo de tecnologías para su análisis en el laboratorio, particularmente de procedimientos diagnósticos y precisos. Existen más de 1500 variantes de hemoglobinas, pero no todas tienen significación clínica, las más relevantes son las talasemias en las que hay un déficit de síntesis de cadenas de globinas y las hemoglobinopatías estructurales, y de ellas la más importante es la Hemoglobina S, drepanocitosis o anemia de células falciformes.4
En 1949, Pauling descubrió que en la anemia falciforme había una alteración en la molécula de hemoglobina; esta enfermedad se encuentra con frecuencia en personas de raza negra y su mestizaje.3
Es la hemoglobinopatía estructural más frecuente y causada por la sustitución de un aminoácido (ácido glutámico) con carga negativa por otro de carga neutra (valina), originada a su vez por la sustitución de la base timina por adenina en el sexto codón del gen de la β-globina, localizado en el cromosoma 11.1
Puede expresarse en cuatro formas clínicas diferentes:
- Forma homocigota o anemia falciforme (HbSS): La mutación afecta a los 2 alelos del gen correspondiente a la cadena β (βs βs). El paciente presenta un 75-95% de HbS, y un 5-15% HbF, presenta graves síntomas clínicos.
- Forma doble heterocigota HbS-Talasemia (HbS-Tal): En el mismo paciente coexisten 2 alelos anormales, uno para la HbS y otro para β-talasemia (βs/ βtal). Si la síntesis a nivel del gen talasémico es nula (β0-tal) la cantidad de HbS será prácticamente la misma que en estado homocigoto (70-90%). Si por el contrario sólo presenta una disminución en el gen talasémico (β+-tal) se observa la existencia de HbA (10-30%), HbS (60-85%), y una pequeña proporción de HbF (5%). También son formas graves, aunque en general no tanto como las formas homocigotas y predominan en el área mediterránea más que en la raza negra.
- Forma doble heterocigota HbS-HbC (HbSC): Coexisten 2 alelos anormales, uno codifica la síntesis de HbS y el otro la síntesis de HbC (βs/ βc). No existe HbA, existen cantidades similares de HbS y HbC (50%). La expresión clínica suele ser menos grave.
- Forma heterocigota o rasgo falciforme (HbAS): La mutación afecta a uno solo de los alelos (βA βS). El paciente tiene un 30-40% de HbS y suele no presentar manifestaciones clínicas.6
Como consecuencia de la mutación, cuando la hemoglobina se desoxigena, sufre un proceso espontáneo de polimerización formando un gel cristalino. Cada polímero está formado por 14 haces longitudinales de deoxi-Hb que se disponen formando un cuerpo tactoide, estructura cilíndrica insoluble y rígida. Debido a estos polímeros se incrementa la rigidez celular y se distorsiona la membrana del eritrocito, adoptando este la forma característica del drepanocito.5
Estos hematíes deformados no pueden atravesar normalmente la microcirculación de los tejidos, son hemolizados y eliminados por los macrófagos del sistema mononuclear fagocítico.
Además de su escasa deformabilidad, produce aumento de la viscosidad sanguínea, facilita la formación de microtrombos y la oclusión de los pequeños vasos; en el proceso de la oclusión vascular también tiene un papel importante la adhesión de los drepanocitos al endotelio vascular lesionándolo y provocando un desequilibro entre vasodilatación y vasoconstricción a favor de esta, los granulocitos liberando citosinas, las plaquetas trombospondina y los reticulocitos presentando ligandos de adhesión facilitando este proceso.6
Las manifestaciones clínicas de la drepanocitosis son consecuencia de la falta de entrega de oxígeno de los eritrocitos falciformes a los tejidos ocasionando hipoxia tisular.2
Objetivo
Identificar de manera oportuna en personas portadoras la presencia de hemoglobina S, para evitar la unión entre los mismos y esto lleve como consecuencia la procreación de hijos homocigotos con anemia de células falciformes, provocándoles principalmente anemia hemolítica, entre otros padecimientos.
Metodología
Materiales a utilizar:
- Portaobjetos
- Cubreobjetos
- Aplicador
- Parafina fundida
Muestra biológica:
Sangre total con anticoagulante EDTA
Reactivo:
Se prepara una solución de metabisulfito de sodio al 2% con 0.5 ml de agua destilada y 0.05 g de metabisulfito de sodio (Na2S2O5). Es de suma importancia que se prepare al momento del que se vaya a utilizar.
Técnica:
En el portaobjetos se colocará 2 gotas del metabisulfito de sodio al 2%, después se agregó una gota de sangre total con EDTA y fueron previamente mezclados con el aplicador; una vez realizado el procedimiento, tomar una pequeña porción con la orilla del cubreobjetos y se colocará con mucho cuidado sobre el portaobjetos para evitar la formación de burbujas.
Se sellará las orillas del cubreobjetos con la parafina fundida y posteriormente se observará a los 15, 30, 60, 120 minutos y 24 horas.
Resultados
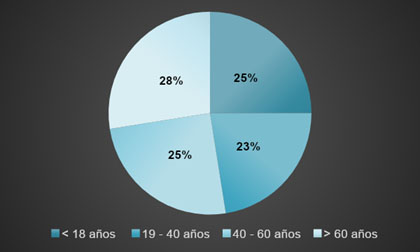
Figura 1. Presencia de hemoglobina S en portadores de acuerdo a edad.
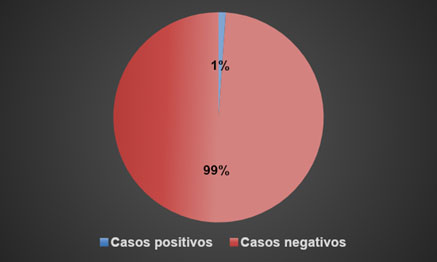
Figura 2. Casos positivos a inducción de drepanocitos en el periodo de marzo-septiembre 2021.
Discusión
La dispersión geográfica del gen βS no permaneció circunscrita al África, sino que se extendió por lo menos a partes circundantes del Mediterráneo.
Durante la época colonial el Puerto de Veracruz fue uno de los principales puntos de entrada al comercio y a miles de esclavos robados de África para su posterior venta, de los cuales se obtuvo un mestizaje de indígena-caucásico africano, heredando así el defecto de hemoglobina S en Veracruz, siendo así que pudiera manifestarse aún en su descendencia.
Conclusión
1. La prevalencia en Centro Diagnóstico Hatver del rasgo drepanocítico, lleva a considerar la importancia de que la inducción de drepanocitos deberá ser incluida en la biometría hemática de manera rutinaria y la electroforesis de hemoglobina en caso de que presente inducción de drepanocitos positiva.
2. Detectar pacientes con rasgo drepanocítico ayudará a evitar o mejorar el manejo clínico de pacientes homocigotos los cuales presentan la enfermedad severa por anemia, hemólisis y crisis vasooclusivas.
Agradecimientos
Al químico David Augusto Hatchett Arenas, co-autor del trabajo por brindar su confianza para la elaboración de este proyecto y permitir tener el acceso a la información que aquí se presenta.
A la química Edith Monserrat García Carrera, co-autora del presente por su asesoría para el desarrollo del trabajo escrito y la interpretación de los resultados.
A mi familia por apoyarme siempre en mis proyectos y motivarme a seguir superándome.
A mis hijos por ser mi principal fuente de motivación para mis proyectos.
Referencias
- Oropeza, T., Flores, A.C., Villegas, M.C., Martínez, J.A., Pulido, N., Baeta, M.F., Moreno, N. (2014). Detección de portadores del rasgo drepanocítico en una muestra de población de Macaray y su zona metropolitana. Comunidad y Salud, 12(1): 46-55.
- Sundd, P., Gladwin, M., Novelli, E. (2019). Pathophysiology of Sickle Cell Disease, HHS Public Access, 14: 263-292.
- Rodríguez, W., Saenz, G., Chavez, M. (1998). Haplotipos de la hemoglobina S: importancia epidemiológica, antropológica y clínica. Revista Panamericana de Salud Pública, 3(1): 1-8.
- Villalva, T., Medina, J., Diaz, M., (2020) Detección de variantes de hemoglobina glicosilada en gestantes. Catlab Informa, 111: 1-10.
- Telen, M., Malik, P., Vercelotti, G. (2019). Therapeutic strategies for sickle cell disease: towards a multiagent approach. HHS Public Access, 18(2): 139-158.
- Heredia, C. (2005). Complicaciones pulmonares de la drepanocitosis. Anales de Pediatría, 62(1): 12-17.
Cuarto Congreso Nacional de Tecnología 24, 25 y 26 de noviembre de 2021,
celebrado en formato virtual

D. R. © UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO.
Excepto donde se indique lo contrario esta obra está bajo una licencia Creative Commons Atribución No comercial, No derivada, 4.0 Internacional (CC BY NC ND 4.0 INTERNACIONAL).
https://creativecommons.org/licenses/by-nc-nd/4.0/deed.es

ENTIDAD EDITORA
Facultad de Estudios Superiores Cuautitlán.
Av. Universidad 3000, Universidad Nacional Autónoma de México, C.U., Delegación Coyoacán, C.P. 04510, Ciudad de México.
FORMA SUGERIDA DE CITAR:
Peres-Quintal, J. A., García-Carrera, E. M., y Hatchett-Arenas, D. A. (2021). Incidencia de rasgo drepanocitico en pacientes de centro diagnostico hatver en Boca del Río, Veracruz. MEMORIAS DEL CONGRESO NACIONAL DE TECNOLOGÍA (CONATEC), Año 4, No. 4, septiembre 2021 - agosto 2022. Facultad de Estudios Superiores Cuautitlán. UNAM.
https://tecnicosacademicos.cuautitlan.unam.mx/CongresoTA/memorias2021/mem2021_paper2.html